
Data content
- I. Data summary
- II. Data processing
- 1. Variant standardization
- 2. Regulatory feature analysis
- 3. Other analysis
- III. Reference data
- IV. References
I. Data summary
rVarBase includes regulatory feature annotations of known and novel human variants. Variants' regulatory features were annotated with: chromatin state of the region surrounding variant, regulatory elements overlapped with variant and variant's potential target genes. It also provides optioned extended annotation for variants and traits associated with variant. The data for rVarBase (as of September 15, 2015) and comparison of the current and previous versions are shown in Table 1.
II. Data processing
As shown in Figure 1, data processing in rVarBase includes: variant standardization, regulatory feature analysis, and other analysis.
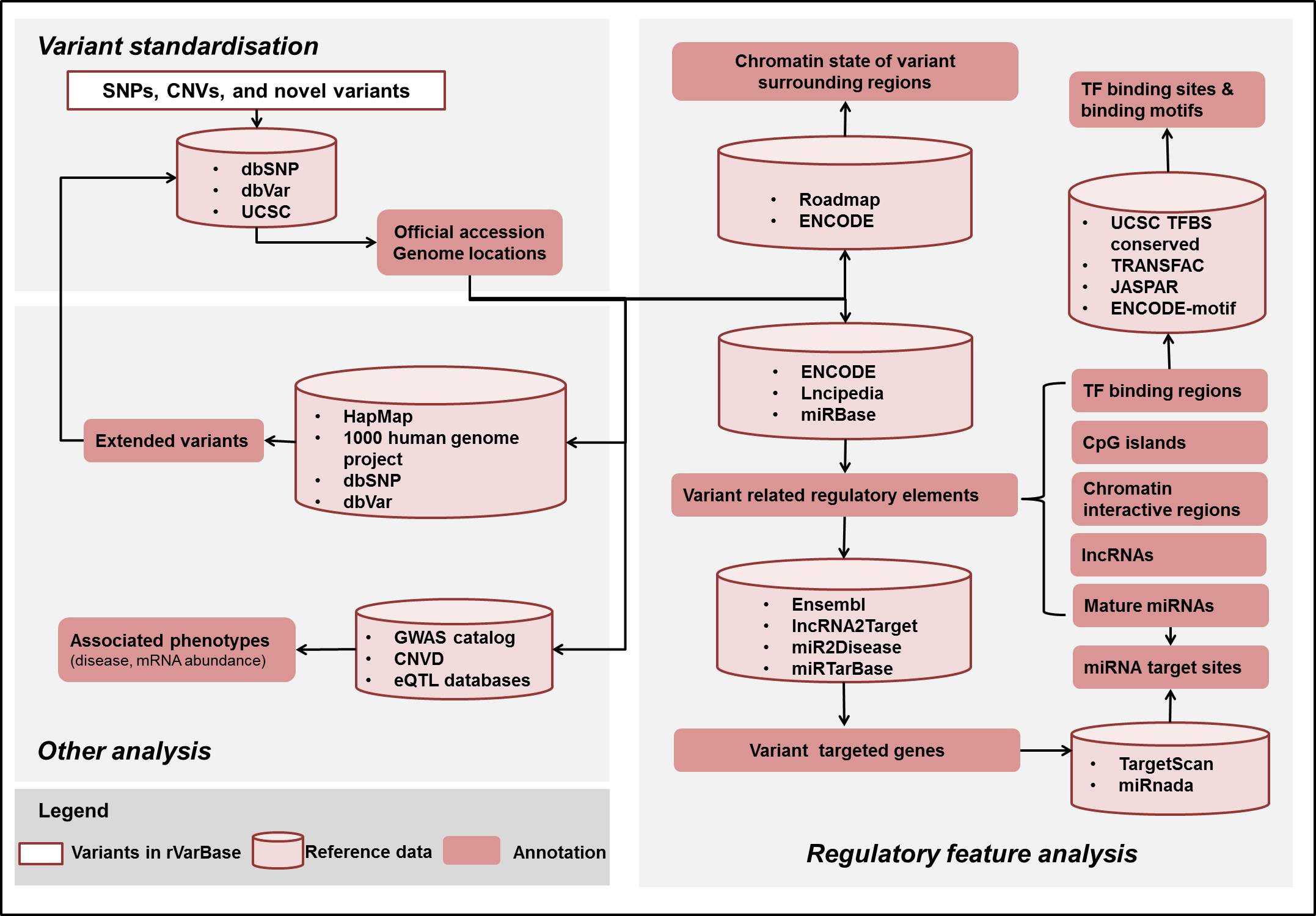
Figure 1 Data processing and data content of rVarBase
1. Variant standardization Official accession and genomic location (with reference to UCSC hg19) of known human variants from dbSNP(version) and dbVar(version) were gotten for subsequent analysis. Novel variants that submitted with their location information were also compared and standardized with information from the two databases.
2. Regulatory feature analysis 1) Chromatin state 8 active states (Active TSS, Flanking Active TSS, Transcr. at gene 5' and 3', Strong transcription, Weak transcription, Genic enhancers, Enhancers, ZNF genes & repeats) and 3 bivalent states (Bivalent/Poised TSS, Flanking Bivalent TSS/Enhancer, Bivalent Enhancer) from the 15-state model that generated by Roadmap final data and ENCODE epigenetic data were utilized to annotate chromatin state of variant¡¯s surrounding region. The detailed chromatin state map was downloaded from the project's supplementary data repository web portal (http://egg2.wustl.edu/roadmap/web_portal/index.html). 2) Variant-related elements obtain and regulation type cataloging. Genomic location of variant was compared with experimentally validated regulatory elements. Elements that covered or overlapped with input variants are identified as variant-related elements. The regulation types that variants involved are cataloged according to their related elements. As shown in Table 1, six types of regulatory elements (CpG island, TF binding region, chromatin interaction region, lncRNA, mature miRNA and miRNA target sites) are taking account in. The potential binding sites of matched TF families inside TF-binding regions were also identified and compared with variants. 3) Regulated gene analysis Variant regulated genes are analyzed according to their related elements. For cis-regulatory elements in transcriptional regulation (CpG island, TF binding sites, chromatin interactive regions) their regulated genes are gained according to their genomic proximity to transcript start sites (TSSs) (within the -5000~+500 region surrounding TSS). For RBP-associated RNA sequences, their target genes are mapped by these RNA sequences. For lncRNA and mirRNA, their regulated genes are obtained from experimentally supported databases.
3. Other analysis 1) Extended variants analysis a. LD-proxies analysis for SNPs. The LD data are compiled from both merged HapMap phases I+II+III genotype data for markers that are up to 200 kb apart and integrated 1000-genomes phase I release data. b. Gaining extended SNP/ CNVs (from dbSNP or dbVar) that overlapped with variants. 2) Associated phenotype analysis a. Variant associated disease are obtained from GWAS catalog and the database of CNVD. b. Variant associated gene expression abundance are required from several eQTL databases and eQTL browser (http://eqtl.uchicago.edu/cgi-bin/gbrowse/eqtl/).
III. Reference data
Reference data of regulatory elements and extended analysis were listed in Table 2 and Table 3 separately .
Table2 Reference data of regulatory elements
IV. References
1. Mathelier, A., Zhao, X., Zhang, A.W., Parcy, F., Worsley-Hunt, R., Arenillas, D.J., Buchman, S., Chen, C.Y., Chou, A., Ienasescu, H. et al. (2014) JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic acids research, 42, D142-147. 2. Kheradpour, P. and Kellis, M. (2014) Systematic discovery and characterization of regulatory motifs in ENCODE TF binding experiments. Nucleic acids research, 42, 2976-2987. 3. Rosenbloom, K.R., Armstrong, J., Barber, G.P., Casper, J., Clawson, H., Diekhans, M., Dreszer, T.R., Fujita, P.A., Guruvadoo, L., Haeussler, M. et al. (2015) The UCSC Genome Browser database: 2015 update. Nucleic acids research, 43, D670-681. 4. Fu, Y. and Weng, Z. (2005) Improvement of TRANSFAC matrices using multiple local alignment of transcription factor binding site sequences. Genome informatics. International Conference on Genome Informatics, 16, 68-72. 5. Volders, P.J., Helsens, K., Wang, X.W., Menten, B., Martens, L., Gevaert, K., Vandesompele, J. and Mestdagh, P. (2013) LNCipedia: a database for annotated human lncRNA transcript sequences and structures. Nucleic acids research, 41, D246-D251. 6. Kozomara, A. and Griffiths-Jones, S. (2014) miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic acids research, 42, D68-D73. 7. Jiang, Q., Wang, Y., Hao, Y., Juan, L., Teng, M., Zhang, X., Li, M., Wang, G. and Liu, Y. (2009) miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic acids research, 37, D98-104. 8. Hsu, S.D., Lin, F.M., Wu, W.Y., Liang, C., Huang, W.C., Chan, W.L., Tsai, W.T., Chen, G.Z., Lee, C.J., Chiu, C.M. et al. (2011) miRTarBase: a database curates experimentally validated microRNA-target interactions. Nucleic acids research, 39, D163-169. 9. Friedman, R.C., Farh, K.K., Burge, C.B. and Bartel, D.P. (2009) Most mammalian mRNAs are conserved targets of microRNAs. Genome research, 19, 92-105. 10. Betel, D., Wilson, M., Gabow, A., Marks, D.S. and Sander, C. (2008) The microRNA.org resource: targets and expression. Nucleic acids research, 36, D149-153. 11. Welter, D., MacArthur, J., Morales, J., Burdett, T., Hall, P., Junkins, H., Klemm, A., Flicek, P., Manolio, T., Hindorff, L. et al. (2014) The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic acids research, 42, D1001-1006. 12. Qiu, F., Xu, Y., Li, K., Li, Z., Liu, Y., DuanMu, H., Zhang, S., Li, Z., Chang, Z., Zhou, Y. et al. (2012) CNVD: text mining-based copy number variation in disease database. Human mutation, 33, E2375-2381. 13. Xia, K., Shabalin, A.A., Huang, S., Madar, V., Zhou, Y.H., Wang, W., Zou, F., Sun, W., Sullivan, P.F. and Wright, F.A. (2012) seeQTL: a searchable database for human eQTLs. Bioinformatics, 28, 451-452. 14. Gamazon, E.R., Zhang, W., Konkashbaev, A., Duan, S., Kistner, E.O., Nicolae, D.L., Dolan, M.E. and Cox, N.J. (2010) SCAN: SNP and copy number annotation. Bioinformatics, 26, 259-262. 15. Cline, M.S., Craft, B., Swatloski, T., Goldman, M., Ma, S., Haussler, D. and Zhu, J. (2013) Exploring TCGA Pan-Cancer data at the UCSC Cancer Genomics Browser. Scientific reports, 3, 2652. 16. Schadt, E.E., Molony, C., Chudin, E., Hao, K., Yang, X., Lum, P.Y., Kasarskis, A., Zhang, B., Wang, S., Suver, C. et al. (2008) Mapping the genetic architecture of gene expression in human liver. PLoS biology, 6, e107. 17. Myers, A.J., Gibbs, J.R., Webster, J.A., Rohrer, K., Zhao, A., Marlowe, L., Kaleem, M., Leung, D., Bryden, L., Nath, P. et al. (2007) A survey of genetic human cortical gene expression. Nature genetics, 39, 1494-1499. 18. Stranger, B.E., Nica, A.C., Forrest, M.S., Dimas, A., Bird, C.P., Beazley, C., Ingle, C.E., Dunning, M., Flicek, P., Koller, D. et al. (2007) Population genomics of human gene expression. Nature genetics, 39, 1217-1224. 19. Barski, A., Cuddapah, S., Cui, K., Roh, T.Y., Schones, D.E., Wang, Z., Wei, G., Chepelev, I. and Zhao, K. (2007) High-resolution profiling of histone methylations in the human genome. Cell, 129, 823-837. 20. Pickrell, J.K., Marioni, J.C., Pai, A.A., Degner, J.F., Engelhardt, B.E., Nkadori, E., Veyrieras, J.B., Stephens, M., Gilad, Y. and Pritchard, J.K. (2010) Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature, 464, 768-772. 21. Montgomery, S.B., Sammeth, M., Gutierrez-Arcelus, M., Lach, R.P., Ingle, C., Nisbett, J., Guigo, R. and Dermitzakis, E.T. (2010) Transcriptome genetics using second generation sequencing in a Caucasian population. Nature, 464, 773-777. 22. Zeller, T., Wild, P., Szymczak, S., Rotival, M., Schillert, A., Castagne, R., Maouche, S., Germain, M., Lackner, K., Rossmann, H. et al. (2010) Genetics and beyond--the transcriptome of human monocytes and disease susceptibility. PloS one, 5, e10693. 23. Ma, B., Huang, J. and Liang, L. (2014) RTeQTL: Real-Time Online Engine for Expression Quantitative Trait Loci Analyses. Database : the journal of biological databases and curation, 2014. 24. Ramasamy, A., Trabzuni, D., Guelfi, S., Varghese, V., Smith, C., Walker, R., De, T., Consortium, U.K.B.E., North American Brain Expression, C., Coin, L. et al. (2014) Genetic variability in the regulation of gene expression in ten regions of the human brain. Nature neuroscience, 17, 1418-1428. 25. Ding, J., Gudjonsson, J.E., Liang, L., Stuart, P.E., Li, Y., Chen, W., Weichenthal, M., Ellinghaus, E., Franke, A., Cookson, W. et al. (2010) Gene expression in skin and lymphoblastoid cells: Refined statistical method reveals extensive overlap in cis-eQTL signals. American journal of human genetics, 87, 779-789. 26. Consortium, G.T. (2015) Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science, 348, 648-660. 27. Abecasis, G.R., Altshuler, D., Auton, A., Brooks, L.D., Durbin, R.M., Gibbs, R.A., Hurles, M.E. and McVean, G.A. (2010) A map of human genome variation from population-scale sequencing. Nature, 467, 1061-1073. 28. Patterson, K. (2011) 1000 genomes: a world of variation. Circulation research, 108, 534-536. 29. Li, Y., Willer, C.J., Ding, J., Scheet, P. and Abecasis, G.R. (2010) MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genetic epidemiology, 34, 816-834.



